The purpose of JACOP is the automated classification of a set of protein sequences.
In contrast with MSA-based/phylogeny approach, JACOP does not require that sequences are arranged as a " meaningful " multiple sequence alignment. JACOP is especially suited for modular proteins.
In addition to a possible classification, JACOP also provides diagnostic clues about the different regions of each sequence in respect to the whole classification (send matches to Catalogue factory).
In contrast with MSA-based/phylogeny approach, JACOP does not require that sequences are arranged as a " meaningful " multiple sequence alignment. JACOP is especially suited for modular proteins.
In addition to a possible classification, JACOP also provides diagnostic clues about the different regions of each sequence in respect to the whole classification (send matches to Catalogue factory).
| Interpretation |
So-called "independent groups" are defined such as no homology is detected for two sequences that belong to two different groups. Within an independent group, the sequences are further partitioned into sub-groups using the PAM (Partitioning Around Medoids) method. The "silhouette coefficient" is used as an indicator of the "quality" of the clustering.
The overall average silhouette width for the entire plot is simply the average of the silhouette coefficient for all objects in the whole dataset. In addition a hierarchical representation (i.e. a tree) of the sequences is also provided to complete the picture even though the classification implied by this dendrogram is less robust than the one produced by the PAM method. Within each independent group, the optimal partitioning is searched for using the PAM method. The minimal and maximal numbers of clusters to evaluate are given according to the vertical gray lines on the tree picture. 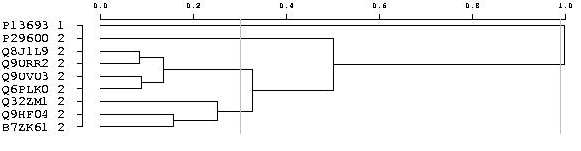 When the number of sequences is too large in an independent group (i.e. more than 200 sequences), the PAM method is not performed. The clusters are obtained by cutting the tree. This partition is less robust and reproducible. The cluster number is evaluated with a single value, chosen by users on the query screen (default is 0.50). A vertical red line highlights this value on the tree picture. 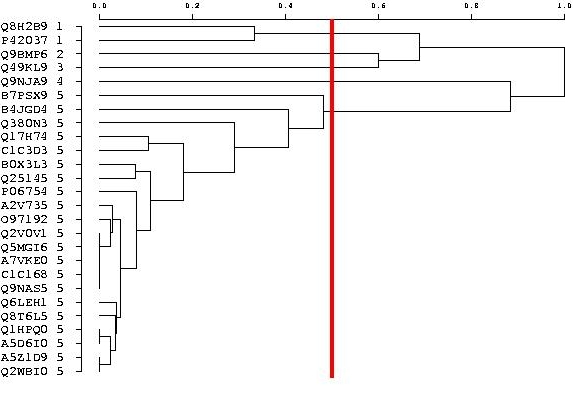
|
JACOP: a simple and robust method for the automated classification of protein sequences with modular architecture.
Sperisen P, Pagni M.
BMC Bioinformatics. 2005 Aug; 6:216. [RIS]
Sperisen P, Pagni M.
BMC Bioinformatics. 2005 Aug; 6:216. [RIS]